

Abstract
Rectovestibular fistula is the most common type of anomaly found in a female newborn with anorectal malformation. However, when the baby is found to have two orifices in the introitus, rectovaginal fistula is much less common and suspected. The rare differential diagnosis of Müllerian agenesis, a condition in which the rectum shifts anteriorly and the vagina is absent, is seldom considered. In many cases, the diagnosis of Müllerian agenesis is made only during definitive anorectoplasty. In view of its impact on management, a proper examination under anaesthesia, imaging studies and a diagnostic laparoscopy may be required to confirm the presence or absence of Müllerian structures in such patients. We herein describe a patient with the rare coexistence of VACTERL association and Müllerian agenesis, and discuss the management of anorectal malformations in female patients with Müllerian agenesis.
INTRODUCTION
Müllerian agenesis is a congenital malformation of the genital tract. While it can exist in isolation or be associated with other congenital anomalies, its coexistence in a patient with VACTERL association is rare. Nevertheless, it is imperative for an accurate diagnosis to be made through detailed and accurate examination in view of the implications for subsequent therapeutic options. Herein, we describe a patient with the rare coexistence of VACTERL association and Müllerian agenesis, and discuss the management of anorectal malformations in female patients with Müllerian agenesis.
CASE REPORT
A female infant was delivered at 38 weeks of gestation with good birth weight. She was found to have oesophageal atresia with a tracheo-oesophageal fistula and underwent a successful repair. Her anal opening was also found to be absent, with passage of meconium noted via an opening in the introitus. Other anomalies suggesting VACTERL association included tethering of the spinal cord with a lipomatous filum, bilateral vesico-ureteric reflux, overactive bladder, a small, scarred right kidney, as well as minor cardiac defects (patent ductus arteriosus, patent foramen ovale and atrial septal defects).
A bedside examination of the perineum suggested the diagnosis of a rectovestibular fistula. The patient was therefore managed initially with washouts via the fistula, which achieved good decompression. Elective posterior sagittal anorectoplasty (PSARP) was carried out at 11 weeks of age. During surgery, the anal opening was found between the labia minora with no apparent vaginal opening. This raised the suspicion of vaginal atresia. The decision was made to proceed with the anorectoplasty. Postoperative ultrasonography of the pelvis could not demonstrate the presence of the uterus. Karyotype cultures, however, confirmed a normal 46 XX genotype.
The patient required management for her other VACTERL association anomalies in the intervening years and received regular follow-up. At five years of age, she presented with repeated episodes of blood stains on her undergarments. Microscopic and macroscopic haematuria as well as bleeding per rectum were ruled out on all occasions. An examination under anaesthesia at that time confirmed the absence of a normal vaginal opening in the introitus (
Fig. 1
Photographs taken during examination conducted under anaesthesia show (a) a single orifice opening into the urethra (arrow); and (b) the insertion of a urinary catheter through the orifice with drainage of urine.
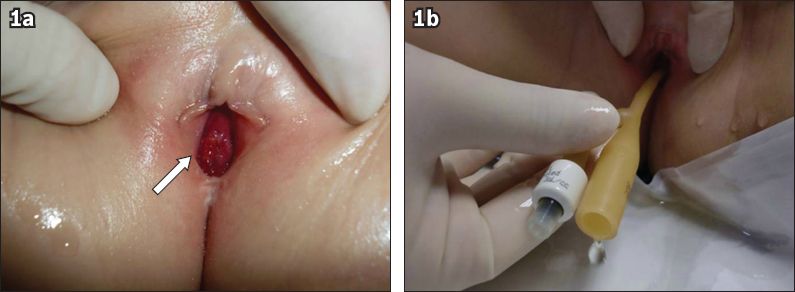
At seven years of age, as part of her evaluation for neurogenic bladder and vesico-ureteric reflux, a cystoscopy was performed. In the same sitting, she underwent diagnostic laparoscopy, which confirmed the absence of a uterus and fallopian tubes (
Fig. 2
Laparoscopic images of the pelvis show (a) an absent uterus; and (b) the presence of ovaries (arrow).
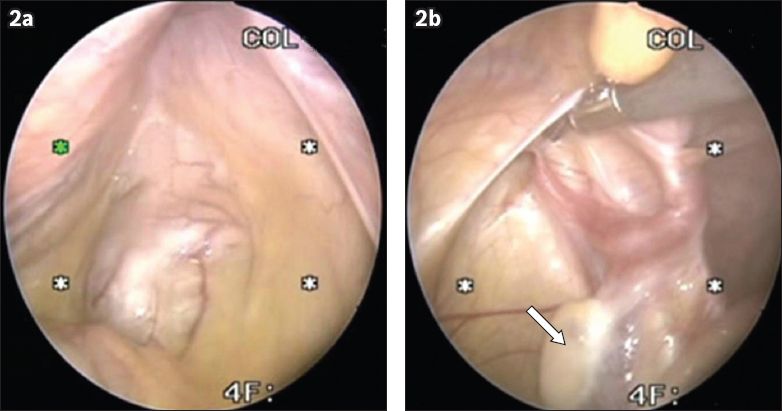
DISCUSSION
Müllerian agenesis is a congenital malformation of the genital tract. The embryological origin of the genital tract is derived initially from the intermediate mesoderm of the urogenital ridge. The Müllerian duct develops as an evagination of the peritoneum adjacent to the gonads and migrates to the cloaca following the Wolffian duct. The distal end of the ducts forms a solid uterovaginal primordium, which contacts the endodermal urogenital sinus at the Müllerian tubercle. Proliferation of the endoderm of the tubercle produces the vaginal plate, which forms the vaginal lumen by canalisation and elongates caudally to bring the opening of the vagina to the exterior of the urogenital sinus. Müllerian agenesis results in a congenital absence of the uterus and vagina, and is one of the most common causes of vaginal agenesis.(1,2)
Müllerian agenesis can exist in isolation or be associated with other congenital anomalies such as renal and skeletal anomalies in MURCS association, a distinctive non-random association involving malformations of Müllerian duct aplasia, renal aplasia and cervicothoracic somite dysplasia. Mayer-Rokitansky-Küster-Hauser syndrome, which can be associated with renal or skeletal abnormalities as well as hearing or cardiac defects, was found in this patient. On the other hand, anorectal malformations, which are birth defects involving the development of the lowest portion of the intestinal and urogenital tracts, often feature in VACTERL association. While there are various congenital abnormalities commonly associated with Müllerian agenesis, its coexistence in a patient with VACTERL association is rare. Müllerian agenesis has only been reported as part of the MURCS association in patients with VACTERL association in a few case reports.(3,4)
The association of Müllerian agenesis with anorectal malformation poses a diagnostic challenge to paediatric surgeons, and the diagnosis of Müllerian agenesis in such patients is usually delayed because of a lack of awareness. In the evaluation of a female neonate with an anorectal malformation, the diagnostic algorithm follows examination findings from a detailed perineal examination. However, examination of a neonatal perineum at the bedside when the baby is awake and kicking can be extremely difficult. Often, the actual number of orifices cannot be clearly defined. While one would expect three orifices (urethral, vaginal and rectal fistula) in a classic rectovestibular fistula, definitive identification of all three is not always possible in the ward. As rectovestibular fistula remains the most common anorectal malformation in a female infant, passage of meconium via the introitus is often assumed to be due to the presence of a rectovestibular fistula. At the definitive anorectoplasty, however, the anatomy can be better defined. When only two orifices are seen, the underlying possibilities would be a rectovaginal fistula and, less commonly, Müllerian agenesis. Our patient’s case history clearly demonstrated the diagnostic difficulties associated with this constellation of rare anomalies.
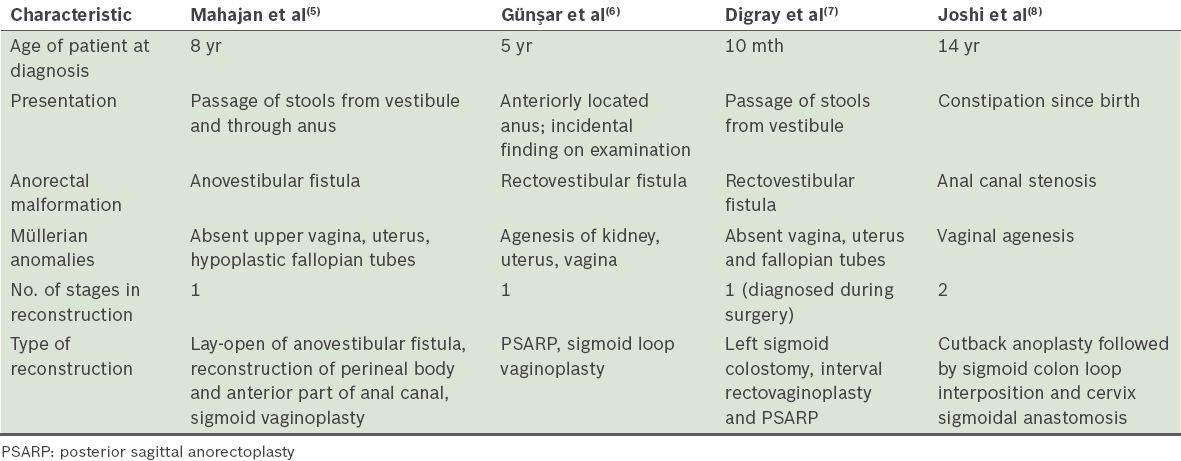
A review of the literature on anorectal malformations that are associated with Müllerian anomalies is summarised in
Table I
Summary of literature on coexistence of Müllerian agenesis and anorectal malformations.
The association of Müllerian agenesis with anorectal malformations has some important treatment implications. Early diagnosis has an impact on eventual clinical management, as the use of the sigmoid colon during a colostomy in the management of the anorectal malformation might preclude its use for vaginoplasty at a later stage. For the same reason, care should be exercised during the anorectoplasty to preserve the integrity of the distal colon. Müllerian agenesis also has serious implications on the child’s eventual ability to reproduce. Early diagnosis of this condition allows the psychological issues and concerns to be carefully evaluated and addressed earlier in the child’s life.(10)
To establish an accurate diagnosis, a female neonate who is found to have two orifices in the introitus on perineal examination should have further evaluation of the genitourinary tract by ultrasonography. However, pelvic ultrasonography performed in early infancy poses many diagnostic challenges, as infants often have many gas-filled bowel loops obscuring accurate visualisation of the pelvic organs. More definitive imaging is often required to evaluate the pelvic organs. In this patient, MR imaging of the pelvis was also advocated to study the genitourinary tract in greater detail. While MR imaging may provide a less invasive option for delineating the anatomy of the Müllerian structures, this imaging modality almost always requires general anaesthesia in the paediatric population. In addition, reconstructed images of the pelvis and perineum are extremely difficult to interpret in prepubertal girls. Thus, if the uterus cannot be visualised on repeated ultrasonography of the pelvis, we would propose an early diagnostic laparoscopy to evaluate for the presence or absence of Müllerian structures so that a definitive diagnosis of Müllerian agenesis can be made.
In conclusion, although the coexistence of Müllerian agenesis and VACTERL association is rare, one must actively check for this possibility in every female child presenting with two orifices in the perineum. In the evaluation of a female neonate with anorectal malformation, it is therefore imperative to have a detailed and accurate examination of the perineum. If there are indeed only two orifices in the introitus, ultrasonographic studies of the pelvis should be performed to localise the uterus, if any. Although it is extremely rare, the concomitant diagnosis of Müllerian agenesis in a child with VACTERL association can have implications for subsequent therapeutic options.