

Abstract
INTRODUCTION
Herpes simplex virus type 2 (HSV-2) is the most common cause of genital herpes. Glycoprotein G (gG) is a prototype antigen for type-specific serodiagnosis distinguishing between HSV type 1 (HSV-1) and HSV-2 infections. As immunological diagnosis kits for accurate differentiation between HSV-1 and HSV-2 antibodies can be expensive, there is a need to develop a convenient, sensitive, specific and cost-effective serodiagnostic kit.
METHODS
We successfully expressed a fragment of gG comprising residues 321–580 of HSV-2 with histidine tag (gG321–580His) in a Bac-to-Bac baculovirus expression system, which had an antigenicity similar to its native counterpart. An indirect enzyme-linked immunosorbent assay (ELISA) was developed using gG321–580His as the diagnostic antigen and evaluated by comparison with a commercial HerpeSelect 2 ELISA immunoglobulin G kit as reference.
RESULTS
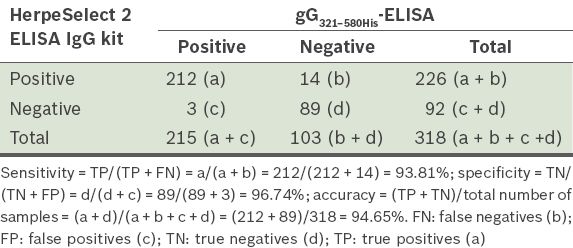
In testing 318 field serum samples, the diagnostic relative sensitivity and specificity of the developed gG321–580His-ELISA test in qualitative comparison with the commercial kit were 93.81% and 96.74%, respectively, and the accuracy was 94.65%.
CONCLUSION
The study indicates that gG321–580His has a high diagnostic potential for HSV-2 virus serodiagnosis in humans.
INTRODUCTION
Herpes simplex virus (HSV) is one of the most common human pathogens. Based on the antigenicity of HSVs, the viruses can be divided into HSV type 1 (HSV-1) and HSV type 2 (HSV-2); both types share a high degree of genetic homology.(1) HSV-1 is the most common cause of oral herpes, while HSV-2 is the most common cause of genital herpes, which is one of the most common sexually transmitted diseases (STDs). Recent studies have shown that HSV-2 infection is related to the occurrence of cervical cancer and that it increases the risk of AIDS infection.(2)
In the United States, genital herpes has reached epidemic proportions, with more than 25% of the adult population infected with HSV.(3) Epidemiological studies indicate that the prevalence of HSV-2 is increasing significantly in China.(4) However, as the majority of individuals infected with HSV-2 have no clinical symptoms, their sexual partners face a substantial risk of infection.(5,6) In addition, primary infection during pregnancy has been associated with spontaneous abortion, prematurity and neonatal herpes.(7) Therefore, the ability to differentiate HSV-2 infection from HSV-1 infection is becoming increasingly important, as it will enable the optimisation of disease treatment as well as help prevent disease transmission to sexual partners and neonates, since suitable precautions can be taken once the disease is identified.
A diagnosis of HSV-2 infection can be achieved using various methods, including virus isolation, nucleic acid techniques, and detection of viral antigens and/or specific antibodies. Type-specific antibody testing for HSV is the most commonly used tool for identifying HSV-2-infected individuals, as it is able to identify subclinical HSV-2 infection and HSV-2-infected individuals who are asymptomatic.(8-10) This method has been used in epidemiological studies, HSV-2 infection diagnosis, and the screening of pregnant women and potential semen donors (the latter is crucial in reducing the risk of HSV-2 transmission).(11-13) While Western blot is considered the most reliable tool for distinguishing between HSV-1 and HSV-2 antibody populations, the procedure is time-consuming and unsuitable for general screening purposes. Enzyme-linked immunosorbent assay (ELISA) is a faster technique with a higher capacity. However, HSV-1 and HSV-2 infections elicit extensive serological cross-reactivity due to their high degree of genetic similarity, which makes it difficult to use serum antibody tests for HSV classification.
Although most HSV antigens are unsuitable as type-specific antigens, the viral envelope glycoprotein G (gG) has been recognised to be suitable; it is the most accepted and widely used type-specific antigen in HSV-2-based seroassays.(14-16) The detection of HSV-2 gG (gG-2) antibodies has been deemed to be a reliable indicator of past or present HSV-2 infection. However, the reliability of using intact gG-2 as a type-specific antigen has recently been questioned because there have been reports that the serostatus of some individuals with definite HSV-2 infection changed from seropositive to seronegative when gG-2 was used as the antigen.(17) Research has shown that the immunodominant epitope area of gG-2 is located at the carboxyl terminal of the protein and that there is no discernible homology between HSV-1 gG (gG-1) and residues 20–546 of gG-2.(16,18) Furthermore, the use of polypeptides as antigens has been shown to result in better specificity and antigenicity than the use of whole proteins.(19)
The significance of HSV type-specific serology is not yet well recognised in China. Commercially available homemade antibody tests, which are based on the whole antigen extract, are not able to accurately distinguish between HSV-1 and HSV-2 antibodies, while reliable immunological diagnosis of HSV-2 depends on expensive imported reagents. This results in some limitations to clinical diagnosis and large-scale epidemiological studies in China. Thus, there is an urgent need for a convenient, high-quality, rapid and inexpensive domestic serodiagnostic kit.
Recombinant proteins can be efficiently produced at reasonable quantities and purity using the baculovirus expression system. The advantages of using the system are that it has the capacity for multiple foreign gene expression and the purification process is relatively simple. The system also has more potential advantages (in terms of immunogenicity, safety and perfect post-translational processing system) than expression systems that use bacteria, yeast or mammalian cells.(20,21)
In our attempt to clone the complete sequence of gG-2 using the baculovirus expression system, we only managed to express a small amount of gG-2. Therefore, we decided to clone a smaller fragment of the gG-2 sequence, comprising residues 321–580. This fragment contains most of the known unique type 2-specific epitopes of gG-2. In the present study, we reported for the first time the production and purification of recombinant HSV-2 virus antigen gG321–580 with histidine tag (gG321–580His) using a baculovirus expression system. The performance of the purified gG321–580His as a coating antigen in indirect ELISA (I-ELISA) was evaluated using a large panel of positive and negative sera.
METHODS
The Bac-to-Bac expression system (Invitrogen, Carlsbad, CA, USA) and Cellfectin reagent (Invitrogen) were used to produce the recombinant fusion protein gG321–580His in Spodoptera frugiperda (Sf9) cell lines. The Sf9 cells and Vero cells used to produce the recombinant fusion protein gG321–580His were cultured in our laboratory – Sf9 cells were propagated in SF-900 II SFM (Gibco, Grand Island, NY, USA) at 27°C and Vero cells were cultured in minimum essential medium at 37°C with 5% CO2. All media contained 10% inactivated fetal bovine sera (FBS) (Gibco), 100 µg/mL streptomycin and 100 IU/mL penicillin.
The HSV-2 viral strain used was isolated from the genitourinary lesions of clinical specimens, which were from patients who presented with genital ulcers that were confirmed to be HSV-2 infections by type-specific polymerase chain reaction (PCR) assays.(22) Virus stocks were prepared by infecting Vero cells. When the cytopathogenic effect was obvious, the cells were collected, washed and suspended in phosphate-buffered saline (PBS). The HSV-2 virus was then released by sonication and the lysate was centrifuged at 1,500 grams for 20 minutes. Aliquots of the virus were stored at –70°C and a single batch was used throughout the study.
Mouse anti-gG-2 monoclonal antibody (Meridian Life Science, Memphis, TN, USA), goat anti-mouse immunoglobulin G (IgG) conjugated to horseradish peroxidase (HRP) (Roche, Nutley, NJ, USA) and HerpeSelect 2 ELISA IgG kit (Focus Diagnostics, Cypress, CA, USA) were used. The MiniBEST Viral RNA/DNA Extraction Kit and all the restriction enzymes, protein molecular weight markers and DNA markers used were from TaKaRa Biotechnology, Dalian, China. All other chemical reagents used were of analytical purity and from commercial sources.
We obtained a total of 318 serum samples, collected from patients with HSV-2 infections, from the STD clinic of the Third People’s Hospital of Hangzhou, China. Of these 318 serum samples, 205 came from male patients (mean age 35.9 ± 4.52 years) and 113 came from female patients (mean age 30.7 ± 4.65 years). Informed consent was obtained from the patients. A total of 20 known HSV-1-positive-HSV-2-negative human sera and ten known HSV-negative human sera were conserved in our laboratory. The sera were frozen and stored at –70°C until HSV-2 antibody testing was performed.
With regard to the construction of recombinant plasmid pFastBac HTc-gG321–580His, we cultivated the HSV-2 virus using well-grown Vero cells. HSV-2 DNA was extracted from the obtained virus culture supernatants, according to the instructions of the MiniBEST Viral RNA/DNA Extraction Kit. Two specific oligonucleotide PCR primers were designed using the gG321–580 gene sequence of gG-2 (GenBank Accession No. NC-001798): (a) upstream primer: 5’-GG
PCR was performed using gG321–580-specific primers, TaKaRa LA Taq polymerase and GC Buffer II. The conditions used were as follows: DNA denaturation at 95°C for 2 minutes; 30 cycles of 95°C for 30 seconds, 50°C for 30 seconds, 72°C for 1 minute; final extension at 72°C for 10 minutes; and save at 4°C. The PCR product was purified using a Gel Extraction Kit (Qiagen, Düsseldorf, Germany) and identified using 1% agarose gel electrophoresis. The purified PCR product was ligated with the pMD18T vector and transformed into DH5α-competent cells. To confirm gG321–580 gene insertion, the pMD18T-gG321–580 vector was purified and digested with BamHI/HindIII. The pFasBac HTc donor plasmid and pMD18T-gG321–580 were prepared via digestion with restriction enzymes BamHI and HindIII. The fragments of interest were purified and recovered from the gel using the Clontech DNA purification system (Clontech, Mountain View, CA, USA). After ligation using T4 DNA ligase, the ligation mixture was transformed into DH5α-competent cells. The recombinant plasmid was identified using restriction endonuclease digestion. Positive clone strains were sequenced and verified by Shanghai Sangon Biotechnology. The resulting recombinant-transposition plasmid was named pFastBac HTc-gG321–580His.
The purified recombinant pFastBac HTc-gG321–580His plasmids were used to transform MAX Efficiency DH10Bac-competent cells, according to the manufacturer’s instructions (Invitrogen). The gG321–580 gene was transposed into Bacmid through lacZ gene disruption. White clones containing the recombinant Bacmids were selected on lysogeny broth agar plates containing 50 µg/mL kanamycin, 7 µg/mL gentamicin, 10 µg/mL tetracycline, 100 µg/mL Bluo-gal and 40 µg/mL isopropyl b-D-1-thiogalactopyranoside. After 36 hours of incubation at 37°C, high-molecular-weight DNA was isolated from the overnight cultures, as described in the manual of the Bac-to-Bac expression system. Successful transposition into the recombinant Bacmid was confirmed using PCR with M13, gG321–580-specific primers.
The mini preparations of recombinant Bacmid DNA were transfected into Sf9 insect cells using the Cellfectin reagent, according to the manufacturer’s instructions. For each transfection, 9 × 105 cells were seeded in a 6-well plate and allowed to attach for at least 1 hour. To form lipid-DNA complexes, we mixed 6 µL of Cellfectin lipid reagent and 1 µg of Bacmid DNA, both of which were diluted separately using 100 µL of Grace’s medium, without antibiotics and FBS. The lipid-DNA mixture was incubated for 20 minutes at room temperature, after which 0.8 mL of Grace’s unsupplemented medium was added into the mixture. The resulting lipid-DNA complex was then laid over the washed Sf9 cells and removed after 5 hours of incubation at 27°C. Then, 2 mL of Grace’s medium supplemented with 10% FBS was added to the plates. The Sf9 cells were incubated in a humidified incubator at 27°C for about three days. Recombinant baculovirus was harvested from the supernatant and stored at 4°C. The virus stock was further subjected to two rounds of amplifications (by infecting fresh Sf9 cells). This was done in order to obtain a high-titre working virus stock. Virus titre was determined by performing a viral plaque assay (Invitrogen), according to the manufacturer’s instruction. The resulting virus titre was stored in aliquots at −80°C until needed.
To express recombinant gG321–580His, 1 × 107 Sf9 cells were infected with the recombinant baculovirus at a multiplicity of infection (MOI) of 5. Infected cells were harvested from the cultures 72 hours after infection, and sonicated for 10 seconds at 50% ultrasonic power with five repeats and a 3-minute period of cooling down on ice. The supernatant was cleared using low-speed centrifugation, which spun down the cell debris. Since the expressed recombinant protein contained 6 × histidine tag at the N-terminal, this ensured effective one-step purification of the recombinant gG321–580His on Ni2+-NTA (nitrilotriacetic acid) resin (Qiagen), which was done according to the manufacturer’s instructions. The supernatant was incubated with the pre-equilibrated (50 mM sodium phosphate, pH 8.0, 300 mM sodium chloride and 10 mM imidazole) Ni2+-NTA resin column. The gG321–580His protein was eluted using an elution buffer that was supplemented with 300 mM imidazole. The eluate was collected in 1 mL fractions, and the eluted fractions before and after pooling were analysed using sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). The eluate was dialysed against demineralised water, and the dialysed culture media containing gG321–580His was concentrated by lyophilisation.
The recombinant gG321–580His was analysed using 12% SDS-PAGE and Western blot. Proteins separated by the 12% SDS-PAGE were transferred onto a nitrocellulose membrane (Amersham, Piscataway, NJ, USA). The membrane was blocked at 4°C with 5% nonfat dried milk in 10 mM Tris-hydrochloric acid (pH 8.0), 150 mM sodium chloride and 0.1% Tween 20 (v/v) overnight. The blot was rinsed with TST (Tris-saline Tween) buffer and incubated with mouse anti-gG-2 monoclonal antibody (1:500) at room temperature for 1.5 hours. After that, the blot was washed three times with TST buffer and incubated with goat anti-mouse IgG conjugated to HRP antibody (1:1000) for 1 hour. The blots were monitored for colour development in a substrate solution containing 0.5 mg/mL diaminobenzidine and 0.01% H2O2. The concentration of the recombinant protein was determined using a Micro BCA Protein Assay Kit (Pierce, Rockford, IL, USA).
I-ELISA was standardised using checkerboard titration procedures described by Wright et al.(23) The purified gG321–580His was diluted in carbonate-bicarbonate buffer (pH 9.6) to a concentration of 5 µg/mL. Flat-bottomed microplates (IWAKI, Chiba, Japan) were coated with gG321–580His (500 ng/well) overnight at 4°C. The plates were then washed three times with a washing buffer consisting of PBS (pH 7.2) and 0.1% Tween 20. Nonspecific binding sites were blocked with 100 µL/well of PBS + Tween (PBST) (i.e. 3% bovine serum albumin, PBS and 0.05% Tween 20) for 1 hour at 37°C and then washed as previously described. A total of 100 µL of serum diluted in PBST (1:100) was added to each well and the plates were incubated at 37°C for 1 hour. Plates were then washed as previously described and incubated with 100 µL HRP conjugated with goat anti-human IgG antibody (1:1000) for 1 hour at 37°C. Thereafter, the plates were washed again and secondary antibody binding was visualised by adding 100 µL substrate solution (containing 50 mL 0.1 mol/L citric acid, 50 mL 0.2 mol/L NaHPO4, 40 mg o-phenylenediamine and 0.015 mL 30% H2O2) into each well, for 30 minutes at room temperature. The reaction was stopped by adding 50 µL of 2 M H2SO4, and the optical density (OD) was measured at 490 nm against a blank control well that had not received serum and conjugate. Each test serum was tested in duplicate. The standard positive and negative sera controls, and conjugate control, were tested in quadruplicate. I-ELISA performed using the home-made kit was designated as gG321–580His-ELISA.
In the present study, the cut-off value was set to be the mean OD490 value of the 20 known negative sera (confirmed by HerpeSelect 2 ELISA IgG kit and by the absence of any clinical symptoms associated with HSV in the patients) + 3 × standard deviations (SDs). Intra-assay precision was determined by measuring the antibody level of one anti-HSV-2-negative serum and two anti-HSV-2-positive sera (of which one had low antibody concentration and the other had high antibody concentration) 10 × under the same conditions. Inter-assay precision, on the other hand, was determined by measuring the antibody level of one anti-HSV-2-negative serum and two anti-HSV-2-positive sera (of which one had low antibody concentration and the other had high antibody concentration) 10 × under the conditions in which the gG321–580His antigen was produced, for ten different days. Each serum sample was analysed in six parallels. For each serum sample, the coefficient of variation (CV) was calculated as the SD divided by the mean of the optical ELISA reading.
The 318 serum samples collected from patients with HSV-2 infections, 20 known HSV-1-positive-HSV-2-negative human sera and ten known HSV-negative human sera were tested for the presence of HSV-2 IgG antibodies using: (a) the gG321–580His-ELISA developed in the present study; and (b) a commercially available HerpeSelect 2 ELISA IgG kit. The relative sensitivity and specificity were determined by comparison with the well-documented and commercially available HerpeSelect 2 ELISA IgG kit as a standard, which has been approved by the United States Food and Drug Administration (FDA). The HerpeSelect 2 ELISA IgG kit is based on recombinant-produced gG-2. In comparison to Western blot, the HerpeSelect 2 ELISA IgG kit was reported to have a sensitivity of 97.9% and a specificity of 95.4%.(24)
RESULTS
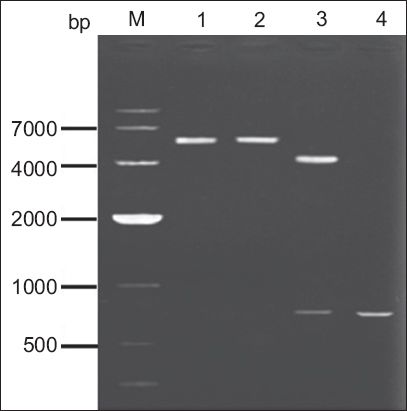
Fragments of the gene gG321–580 were amplified by PCR using specific primers. The resulting sequence of the cDNA produced (GenBank Accession No. KC626072), which was about 800 bp in length, gave 99% homology scores with the known gene sequence of gG321–580 (GenBank Accession No. NC-001798) by sequencing (data not shown). Restriction endonuclease digestion was performed to verify the correct insertion of the gene gG321–580 in the recombinant pFastBac HTc-gG321–580His. Gel electrophoresis in 1% agarose showed gG321–580 (0.8 kb) and pFastBac HTc-gG321–580His plasmid (5.6 kb) (
Fig. 1
Agarose gel electrophoresis of the polymerase chain reaction (PCR) amplification products, for the identification of pFastBac HTc-gG321–580His. Lane M: DNA marker; lane 1: pFastBac HTc-gG321–580His digested by BamHI; lane 2: pFastBac HTc-gG321–580His digested by HindIII; lane 3: pFastBac HTc-gG321–580His digested by BamHI/HindIII; lane 4: PCR products of pFastBac HTc-gG321–580His using gG321–580-specific primers.
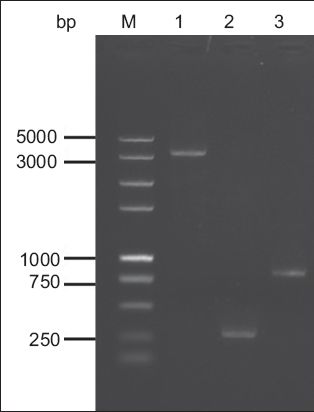
The size of the Bacmid DNA is > 135 kb. As it would be difficult to verify the insertion of the gG321–580 gene in the recombinant Bacmid-gG321–580His using classical restriction endonuclease digestion analysis, PCR was used to confirm the recombinant Bacmid-gG321–580His. The M13(+)/M13(–) amplification primers were directed at sequences on either side of the mini-attTn7 site with the lacZ a-complementary region of the Bacmid. Using M13(+)/M13(–) amplification primers, the amplification products from the recombinant Bacmid-gG321–580His would generate a 3.2 kb band, while amplification of the non-recombinant Bacmid would generate a 300-bp band. Using gG321–580 specific primers, the amplification products from the recombinant Bacmid-gG321–580His would generate a band of about 800 bp. Results indicated that the recombinant baculovirus was constructed successfully (
Fig. 2
Agarose gel electrophoresis of the polymerase chain reaction (PCR) amplification products, for the identification of recombinant Bacmid-gG321–580His. Lane M: DNA marker; lane 1: PCR products of Bacmid-gG321–580His using M13 primers; lane 2: PCR products of wild Bacmid using M13 primers; lane 3: PCR products of Bacmid-gG321–580 His using gG321–580-specific primers.
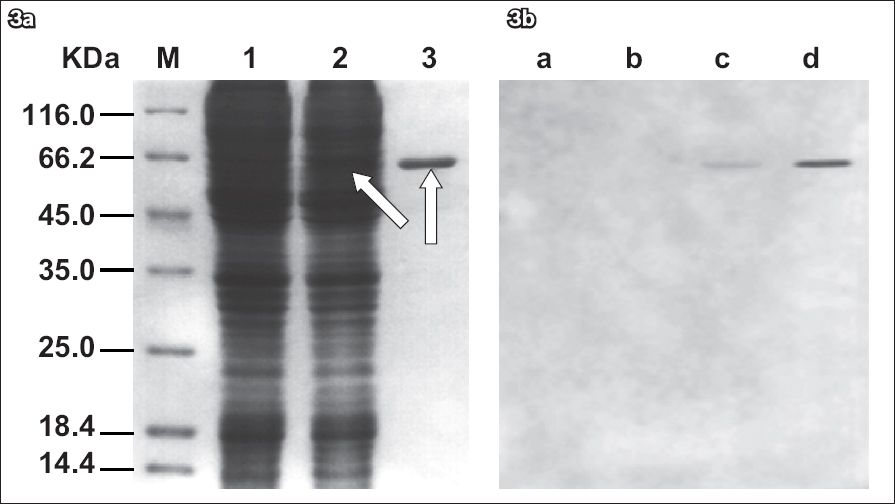
Recombinant Bacmid-gG321–580His was isolated from overnight cultures and transfected into Sf9 cells with Cellfectin reagents. After the infected cells stopped dividing and enlarged with pathological changes, the Bacmid-gG321–580His transfected cell cultures were collected and analysed. To test whether recombinant gG321–580His proteins were expressed, lysates of the Sf9 cells infected with recombinant Bacmid-gG321–580His were analysed using Coomassie blue staining after the proteins had been separated by SDS-PAGE and Western blot. Lysates of the Bacmid-gG321–580His-infected Sf9 cells showed a singular band of about 60 kDa molecular weight. The non-infected Sf9 cells showed no signals (
Fig. 3
(a) SDS-PAGE of the lysates of the non-infected Sf9 cells and the baculovirus-infected Sf9 cells. Lane 1: lysates of the non-infected Sf9 cells; lane 2: lysates of the baculovirus-infected Sf9 cells; lane 3: purified recombinant gG321–580His. The lysates were separated using 12% SDS-PAGE. Additional bands (arrows) were observed in lanes 2 & 3 after Coomassie blue staining. A protein marker is shown at the left part of the Coomassie-stained gel (lane M). (b) Western blot analysis of the recombinant gG321–580His. Lane a: protein marker; lane b: non-infected Sf9 cells culture; lane c: baculovirus-infected Sf9 cells culture; lane d: purified recombinant gG321–580His. A single band was observed in both lanes c & d, which showed that the recombinant protein gG321–580His was able to react with the specific mouse anti-gG-2 monoclonal antibody. No Western blot signal was observed in lanes a & b (negative controls), indicating that the recombinant protein gG321–580His has similar functions with its natural counterpart.
The recombinant fusion protein gG321–580His was used in I-ELISA for the detection of HSV-2-specific antibodies in sera. Optimal ELISA conditions were determined using checkerboard assays. The optimal antigen concentration for discrimination between HSV-2 positive and negative reactions was found to be 500 ng/well, and the optimal serum dilution was found to be 1:100.
In the present study, the cut-off value was set at 0.38, based on the mean OD490 of the 20 negative controls. The intra- and inter-assay precisions of each ELISA, using the one anti-HSV-2-negative serum and the two anti-HSV-2-positive sera (of which one had low antibody concentration and the other had high antibody concentration), were established to give preliminary evidence of the repeatability of each assay. The gG321–580His-ELISA showed good repeatability with an intra-assay CV of 4.8% and an inter-assay CV of 9.3%.
Sensitivity, specificity and accuracy were calculated by comparing the developed gG321–580His-ELISA with the commercial HerpeSelect 2 ELISA IgG kit. The performance of the developed gG321–580His-ELISA in identifying the 318 sera collected from patients with HSV-2 infections is shown in
Table I
Relative sensitivity, specificity and accuracy of the gG321–580His-ELISA, when compared with HerpeSelect 2 ELISA IgG kit results, using 318 sera.
DISCUSSION
HSV-1 and HSV-2 are two closely related viruses that infect humans. Both viruses produce orogenital lesions, and can infect the eye, skin and nervous system. Following primary infection, the virus can enter a latent state in neurological tissue and periodically reactivate to produce recurrent infections. Reactivations are frequently asymptomatic, and it is in the absence of overt clinical lesions that the viruses are often shed and transmitted.(25) It is important to distinguish HSV-2 infections from HSV-1 infections, as this would enable the reduction of sexually and vertically transmitted HSV-2 infection, as well as the optimisation of treatment for patients with genital herpes infections.(26)
Diagnostic methods include virus isolation, PCR and detection of type-specific HSV antibodies. Virus isolation is fastidious, laborious, time-consuming and costly. Although PCR is very sensitive, the technique has the disadvantage of being highly susceptible to PCR carry-over contamination errors, which can potentially lead to false-positive results.(27) Western blot is considered to be the gold standard for the detection of HSV-1- and HSV-2-specific antibodies, as it has both high sensitivity and high specificity. However, the method is unsuitable for routine diagnostic purposes, as it is relatively expensive, laborious, time-consuming and complex to perform. Furthermore, the results of Western blots are difficult to interpret.
The use of ELISA for the detection of HSV-1- and HSV-2-specific antibodies potentially resolves the problem of inadequate diagnostic performance and lack of standardisation. It is also relatively more rapid and cost-efficient to be performed in typical laboratories. However, purification of the virus is laborious and expensive, and the use of semi-purified or unpurified whole viral antigens in ELISA results in unspecific signal.(28) In contrast, the use of recombinant antigens provides an inexpensive, abundant and defined source of antigen, which would enable the development of highly standardised tests. In addition, ELISA kits that are based on recombinant antigens lack infectivity and cross-reactivity, are very stable and easy to optimise, and do not require the cultivation of the virus (making them suitable for wide distribution).(29,30) Recombinant-based serological assays have been developed for the diagnosis of many viral infections.
Most of the envelope glycoproteins of HSV-1 and HSV-2 have extensive genetic similarities and thus induce a cross-reactive antibody response. The use of type-specific testing based on gG for the diagnosis of genital herpes was recommended by the Centers for Disease Control and Prevention in its guideline for the management of genital herpes.(31) gG-1 and gG-2 contain 238 and 699 amino acids, respectively.(32) In a previous study, the use of phage peptide display library mapping and pepscanning technologies identified a region within the gG-2, located between amino acid residues 351–427 and 525–587, that contains predominantly type 2-specific epitopes.(5) Thus, in the present study, we decided to synthesise a gG-2 peptide that corresponds to the previously recognised immunodominant epitope spanning residues 321–580 of the protein, to use as a multiple antigenic peptide for the type-specific serodiagnosis of HSV-2 (i.e. to detect antibodies from the sera of patients suspected to be infected with HSV-2). In addition to being relatively economical to produce and easy to subject to quality controls and standardisations, recombinant gG321–580His proteins would be safe for use in laboratories that are not equipped with biocontainment facilities. We were interested to test whether recombinant gG321–580 of HSV-2 would be a valuable immunodiagnostic reagent in view of its usefulness in distinguishing HSV-1 from HSV-2.
The Bac-to-Bac baculovirus expression system is a eukaryotic gene expression system that allows rapid and efficient generation of recombinant baculovirus. It is able to produce large amounts of recombinant protein efficiently. The recombinant proteins produced are properly assembled with appropriate folding of glycosylated proteins (essential for biological activities, stability and longevity) and are also easily purified.(33,34)
In the present study, the baculovirus expression system and Sf9 insect cells were used to express large amounts of recombinant gG321–580 with 6 × histidine tag at the N-terminal. The histidine tags helped make the purification procedure even more convenient, as Ni2+-NTA resin could be used to separate the proteins. We infected Sf9 suspension cultures with the recombinant viruses at an MOI of 5 and harvested the media 72 hours after infection. Western blot analyses confirmed successful expression of gG321–580His in the baculovirus-infected insect cells. The gG321–580His produced had similar immunological reactivity as its natural counterpart and molecular weight of approximately 60 kDa; it was larger than predicted, probably due to glycosylation.
Type-specific IgM responses generally do not appear in patients with recurrent HSV-2 genital herpes, whereas IgG antibodies are induced against the gG-2 protein during both primary and recurrent infections. Therefore, we evaluated the performance of the baculovirus-produced recombinant gG321–580His as the coating antigen for the detection of type-specific IgG antibodies against HSV-2 in an I-ELISA. We performed a checkerboard titration with positive and negative human sera to determine the optimal reaction conditions. The OD cut-off values were calculated as the mean results of 20 negative sera + 3 SD. For gG321–580His, antibody detection was found to be optimal at a 1:100 serum dilution and an antigen concentration of 5 µg/mL; the cut-off value of the assay was 0.38.
In the present study, the gG321–580His based assays were found to be reproducible with an intra-assay CV of 4.8% and an inter-assay CV of 9.3%. In addition, the sensitivity and specificity of the gG321–580His-ELISA was evaluated by comparing its results with that of a commercial HerpeSelect 2 ELISA IgG kit. The HerpeSelect 2 ELISA IgG kit, which has been licensed by the United States FDA, is the most commonly used kit for type-specific HSV diagnosis. A panel of 318 serum samples was tested separately using both the commercial ELISA kit and the gG321–580His-ELISA. Qualitative comparison of the results of the gG321–580His-ELISA and the HerpeSelect 2 ELISA IgG kit revealed that the relative sensitivity and specificity of the gG321-580His-ELISA were 93.81% and 96.74%, respectively, and that its accuracy was 94.65%. None of the 20 known HSV-1-positive-HSV-2-negative human sera and none of the ten known HSV-negative sera reacted with gG321–580His-ELISA. Hence, the results of the present study confirm the potential value of the gG321–580His recombinant protein as an antigen in I-ELISA for the detection of specific anti-HSV-2 antibodies. In addition, the use of the home-made I-ELISA may be preferred, as it has similar reliability rates as commercially available kits and is less expensive.
In conclusion, the gG321–580His protein, expressed in a Bac-to-Bac expression system, is a promising novel antigen for the detection of type-specific IgG antibodies when there is suspicion of HSV infection. When the gG321–580His protein was used to develop an I-ELISA for the detection of specific anti-HSV-2 antibodies, the I-ELISA compared well with the commercial HerpeSelect 2 ELISA IgG kit. As the I-ELISA method used in the present study was shown to be sensitive, specific and cost-effective, it may be useful for epidemiological studies that seek to control the spread of HSV-2 infection in developing countries.
ACKNOWLEDGEMENTS
The present study was supported by the Science and Technology Project of Hangzhou (No. 20100733Q28) and the Medical Science and Technology Project of Hangzhou Health Bureau (No. 2010Z010).