

Abstract
INTRODUCTION
Children with Down syndrome (DS) are at increased risk of developing distinctive clonal myeloid disorders, including transient abnormal myelopoiesis (TAM) and myeloid leukaemia of DS (ML-DS). TAM connotes a spontaneously resolving congenital myeloproliferative state observed in 10%–20% of DS newborns. Following varying intervals of apparent remission, a proportion of children with TAM progress to develop ML-DS in early childhood. Therefore, TAM and ML-DS represent a biological continuum. Both disorders are characterised by recurring truncating somatic mutations of the GATA1 gene, which are considered key pathogenetic events.
METHODS
We herein report, to our knowledge, the first observation on the frequency and nature of GATA1 gene mutations in a cohort of Malaysian children with DS-associated TAM (n = 9) and ML-DS (n = 24) encountered successively over a period of five years at a national referral centre.
RESULTS
Of the 29 patients who underwent GATA1 analysis, GATA1 mutations were observed in 15 (51.7%) patients, including 6 (75.0%) out of 8 patients with TAM, and 9 (42.9%) of 21 patients with ML-DS. All identified mutations were located in exon 2 and the majority were sequence-terminating insertions or deletions (66.7%), including several hitherto unreported mutations (12 out of 15).
CONCLUSION
The low frequency of GATA1 mutations in ML-DS patients is unusual and potentially indicates distinctive genomic events in our patient cohort.
INTRODUCTION
Down syndrome (DS) is the most common congenital cytogenetic abnormality, with an incidence of one in 600–800 live births.(1) The syndrome is characterised by recognisable physical features, cognitive disability, and a varying frequency of cardiac, gastrointestinal, skeletal and endocrine defects. DS is also characterised by an increased susceptibility to childhood leukaemia,(2-4) including an increased frequency of distinctive clonal myeloid disorders in early childhood. A spontaneously resolving congenital myeloproliferative state designated as transient abnormal myelopoiesis (TAM) is encountered in 10%–20% of DS newborns.(5) After a period of apparent remission (1–4 years), 20%–30% of DS children with TAM progress to develop a characteristic, treatment-responsive megakaryoblastic form of acute myeloid leukaemia termed myeloid leukaemia of DS (ML-DS).(6,7)
TAM and ML-DS thus represent a continuum of clonal myeloproliferation in DS. Fundamental to both disorders is the gene-dosage imbalance of the human chromosome 21.(8) An additional shared, critical pathogenetic event involves the acquisition of characteristic somatic mutations in the GATA1 gene.(9) The gene, located on chromosome X (Xp11.23), encodes a key haematopoietic transcription factor involved in erythroid and megakaryocyte differentiation. These mutations, involving exons 2 or 3 of the GATA1 gene, result in synthesis of an aberrant truncated isoform (termed short GATA1 or GATA1s) that is putatively oncogenic.(10) The mechanistic basis for somatic GATA1 mutations and the additional molecular events that determine progression from TAM to ML-DS are the focus of intense research.(11,12)
This observational study attempts to characterise, for the first time, to our knowledge, the frequency and nature of somatic GATA1 mutations in DS children with TAM and ML-DS, encountered consecutively over a period of five years at a national paediatric oncology referral centre in Malaysia.
METHODS
From January 2007 to December 2011, children with DS who were consecutively diagnosed to have TAM and/or ML-DS at University Malaya Medical Centre, Kuala Lumpur, Malaysia, were included in the study. The diagnosis of TAM was based on observation of unequivocal blasts on peripheral blood microscopy. ML-DS was confirmed by marrow studies, including microscopy, flow cytometry and, where indicated, bone marrow histology. Parental consent was obtained in all cases. Institutional approval was granted to collect, store and study peripheral blood and/or bone marrow samples from children in the study (University of Malaya Ethical Approval Reference MEC 2008/678.13). Clinical and laboratory data were retrieved from patient medical records.
Mononuclear cells were isolated from peripheral blood (TAM) and bone marrow (ML-DS) by erythrocyte lysis, followed by serial centrifugation. Genomic DNA was extracted from mononuclear cells using standard chloroform-phenol isolation. Screening for GATA1 mutations was carried out by selective stepwise analysis of exons 2 and 3, with exon 3 sequences analysed in samples bearing wild-type exon 2. The exon 2 sequence was amplified using primers (forward 5’-GGAAGGATTTCTGTGTCTGAG-3’ and reverse 5’-GCACTCAGCCAATGCCAAGA-3’) and polymerase chain reaction (PCR) cycling conditions reported by Rainis et al.(13) Exon 3 segments were amplified using primers (forward 5’-GCACTCAGCCAATGCCAAGA-3’ and reverse 5’-GAGCTAGGCTCAGCTCAGCTTTAC-3’) and PCR cycling conditions reported by Nichols et al.(14) All PCR reactions were carried out in 25 µL volumes using the HotStarTaq DNA Polymerase (QIAGEN, Hilden, Germany). PCR products were processed using the QIAquick PCR Purification Kit (QIAGEN, Hilden, Germany) to remove reaction residuals and subjected to automated nucleotide sequencing (Applied Biosystems ABI 3730XL; Thermo Fisher Scientific, Waltham, Massachusetts, USA). PCR amplification was tested with an adequate number of controls. Sequence analysis was performed using the online open-source BioEdit sequence alignment tool (version 7.0.1); the NCBI GATA1 sequence NG_008846.1 served as a reference sequence. All sequence variations and predicted amino acid changes were categorised according to nomenclature recommendations from the Human Genome Variation Society (HGVS).(15)
RESULTS
A total of 33 patients with DS-associated myeloid disorders were encountered over the duration of the study, consisting of nine patients with TAM and 24 patients with ML-DS. Their clinical characteristics and laboratory data are summarised in Tables
Table I
Clinical characteristics and laboratory data of the transient abnormal myelopoiesis (TAM) patients (n = 9).
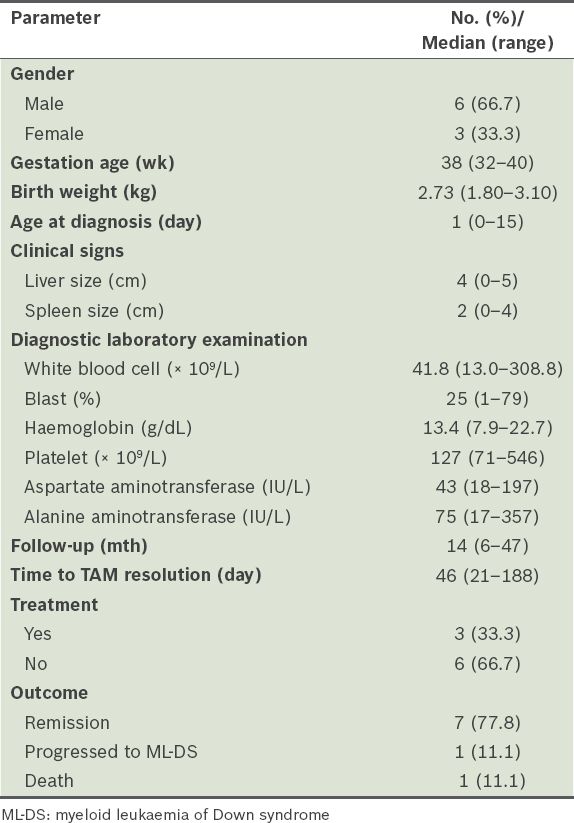
Table II
Clinical characteristics and laboratory data of the myeloid leukaemia of Down syndrome (ML-DS) patients (n = 24).
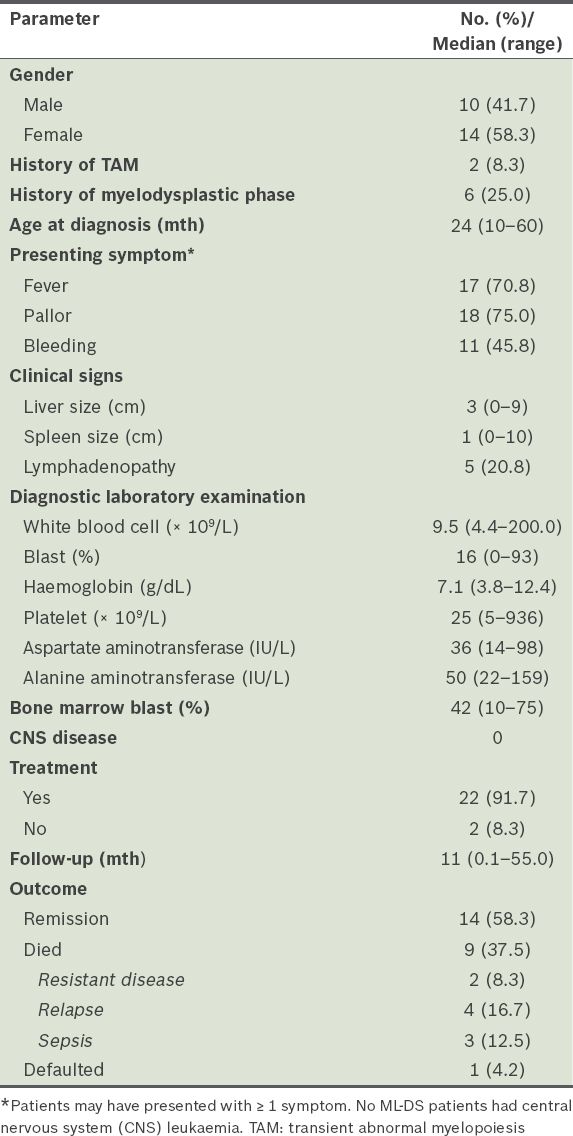
ML-DS was diagnosed at a median of 24 months of age. History of a preceding haematological disorder (two with TAM and six with myelodysplastic phase) was noted in 8 (33.3%) children with ML-DS. Common presenting features included fever, pallor and bleeding. Two children presented with very high leucocyte counts (≥ 100 × 109/L); none had central nervous system leukaemia. 22 (91.7%) children with ML-DS received cytotoxic therapy. The estimated overall five-year survival rate of children with ML-DS was 58.3%; nine children died, including three due to treatment toxicity (i.e. sepsis).
A total of 29 samples were analysed for GATA1 mutations; four samples did not have adequate material for analysis. GATA1 exon 2 mutations were detected in 6 (75.0%) of the eight TAM patients and 9 (42.9%) of the 21 ML-DS patients (
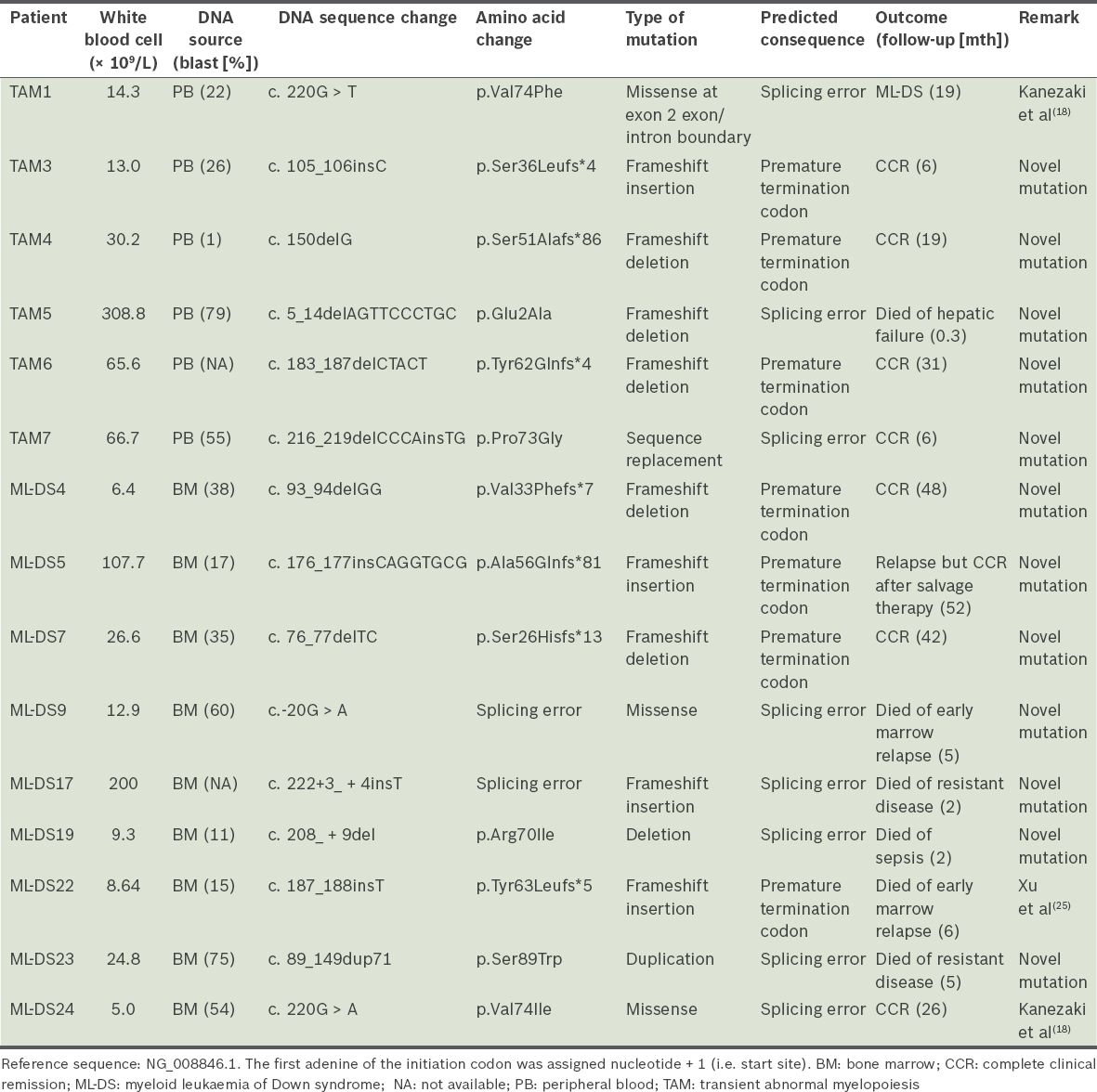
Table III
Characteristics of GATA1 mutations.
DISCUSSION
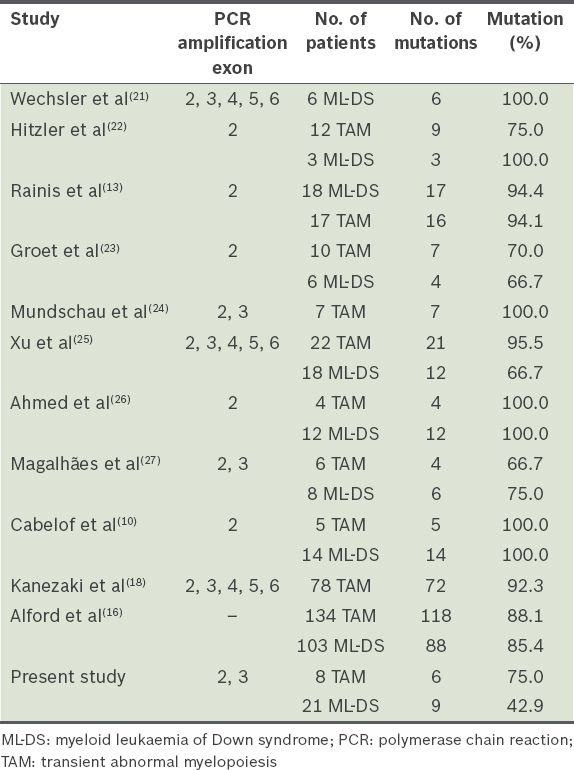
This is the first report of GATA1 mutations in a cohort of Malaysian children with DS-associated myeloid disorders. Although the sample is small, truncating exon 2 GATA1 mutations were encountered in 75.0% of DS neonates who had TAM, a finding that mirrors published observations (
Table IV
Comparison of GATA1 mutation detection rate.
All sequence variations in this study were described in accordance with the nomenclature recommendations of the HGVS.(17) The majority of the exon 2 GATA1 mutations in our cohort were sequence deletions and insertions (66.7%), resulting in frameshift errors and premature termination codons. No exon 3 mutations were identified. 80.0% (n = 12) of observed mutations had not previously been reported and are likely to be novel. However, sequence variations in GATA1 have been inconsistently reported, making it difficult to evaluate GATA1 mutations in our series against those in the published literature. For instance, despite using the same GATA1 reference sequence, Alford et al, who conducted the largest study of GATA1 mutations in DS patients to date, unusually designated the first nucleotide of exon 1 as an indication of the start of the GATA1 sequence.(16) Uniform reporting of sequence variations, in line with standard HGVS terminology, is expected to eliminate such discrepancies in future studies.
Lower relative quantitative expression of the truncated GATA1s protein has been reported to be associated with higher risk of progression to ML-DS.(18) By contrast, the majority of mutations in our series are predicted to be associated with high protein expression of the truncated GATA1s protein (66.7%). In the one patient with TAM who progressed to ML-DS, the mutation resulted in a splicing error predicted to be associated with high expression of GATA1s. High GATA1s expression is also predicted in six of nine mutation-positive ML-DS patients (66.7%). These observations indicate continued uncertainty about the link between the type of GATA1 mutation in TAM and the risk of progression to ML-DS.(16)
Children with ML-DS have been shown to have a superior outcome when compared with children without DS who developed acute myeloid leukaemia (AML). Children with ML-DS experience event-free survival rates of more than 80% as compared with 30%–40% in non-DS children, particularly those with FAB-M7 subtype AML.(19) It was proposed that GATA1 mutations may be important prognostic factors. Acquired GATA1 mutations are postulated to cause decreased transcription and expression of cytidine deaminase (CDA) gene. CDA enzymes catalyse the deamination of cytosine arabinoside (ara-C) to its inactive derivative, ara-U. In the presence of GATA1 mutations, there is a higher concentration of intracellular ara-C, leadings to an increase in cytotoxic efficacy. In our ML-DS cohort, the presence of GATA1 mutations may not necessarily confer a good prognosis. Four out of nine patients with GATA1 mutations developed relapse or resistant disease; one patient from the cohort with negative GATA1 mutation had relapse disease (
Fig. 1
Flowchart shows clinical outcome of children with myeloid leukaemia of Down syndrome (ML-DS). Of the 24 ML-DS patients, 21 had GATA1 analysis performed. 2 patients who died did not have GATA1 analysis performed.
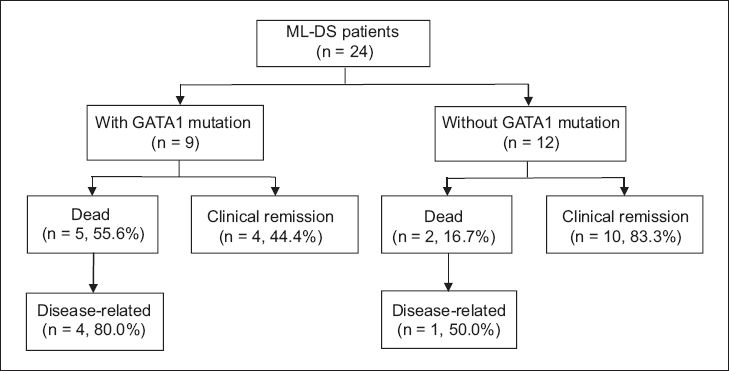
Despite the limitations of sample size, our findings reiterate the observation of recurring GATA1 mutations in children with DS-related myeloid disorders. The high incidence of potentially novel GATA1 mutations and the low frequency of such mutations in exons 2 and 3 raise the prospect of distinctive genomic events in our patient cohort. In our study, the mere presence of GATA1 mutations in ML-DS does not necessarily confer a good prognosis.